A Doença do Xarope de Bordo, também conhecida como leucinose ou MSUD (Maple Syrup Urine Disease — Doença da Urina com Cheiro de Xarope de Bordo), é uma doença genética rara e grave que compromete o metabolismo de aminoácidos essenciais. Essa condição impede o organismo de quebrar corretamente a leucina, isoleucina e valina, levando ao acúmulo de substâncias tóxicas no sangue.
Com base em evidências científicas e prática clínica, reforço que sem diagnóstico e tratamento precoces, a leucinose pode causar danos neurológicos irreversíveis, convulsões, coma e até risco de morte. Felizmente, quando identificada cedo e manejada corretamente, é possível garantir qualidade de vida e desenvolvimento saudável ao paciente.
Neste guia completo, compartilho minha análise técnica e científica, reunindo informações atualizadas sobre causas, sintomas, diagnóstico, tratamento, prognóstico e cuidados essenciais.
O que é a Doença do Xarope de Bordo?
A Doença do Xarope de Bordo é um erro inato do metabolismo, causado por uma mutação genética autossômica recessiva que compromete o funcionamento do complexo enzimático chamado desidrogenase dos cetoácidos de cadeia ramificada (BCKD).
Essa enzima é responsável por degradar três aminoácidos essenciais:
Isoleucina
Valina
Quando o organismo não consegue metabolizar essas substâncias, ocorre o acúmulo de aminoácidos e seus cetoácidos tóxicos, provocando intoxicação metabólica e afetando principalmente o sistema nervoso central.
Por que a doença recebe esse nome?
O nome curioso vem de um dos sinais mais característicos da doença: o odor adocicado da urina, semelhante ao cheiro de xarope de bordo (maple syrup) ou açúcar queimado.
Esse cheiro é causado pelo acúmulo de cetoácidos no organismo, eliminados pela urina, suor e cerúmen (cera do ouvido).
Tipos de Doença do Xarope de Bordo
A leucinose pode se manifestar em diferentes formas clínicas:
1. Forma Clássica
Mais grave e comum
Sintomas surgem nos primeiros dias de vida
Evolução rápida se não tratada
2. Forma Intermediária
Evolução mais lenta
Sintomas mais leves no início
3. Forma Intermitente
Episódios metabólicos ocasionais
Pode se manifestar mais tarde na infância
4. Forma Sensível à Tiamina
Responde parcialmente à suplementação de vitamina B1
Principais Sintomas e Sinais de Alerta
Os sintomas costumam aparecer entre o 3º e o 7º dia de vida, especialmente na forma clássica.
Em recém-nascidos
Odor adocicado da urina e cera do ouvido
Dificuldade para mamar
Baixo apetite
Letargia e sonolência intensa
Choro fraco
Hipotonia (flacidez) ou rigidez muscular
Vômitos
Convulsões
Respiração irregular
Em crianças sem tratamento
Atraso no desenvolvimento neuropsicomotor
Déficits cognitivos
Dificuldades motoras
Problemas de coordenação
Risco de coma em crises metabólicas
O que causa a Doença do Xarope de Bordo?
A doença é causada por mutações genéticas herdadas dos pais, que geralmente são portadores assintomáticos.
Características genéticas
Herança autossômica recessiva
Ambos os pais precisam carregar o gene alterado
Cada gestação tem:
25% de chance da criança ser afetada
50% de chance de ser portadora
25% de chance de não herdar o gene
Diagnóstico da Doença do Xarope de Bordo
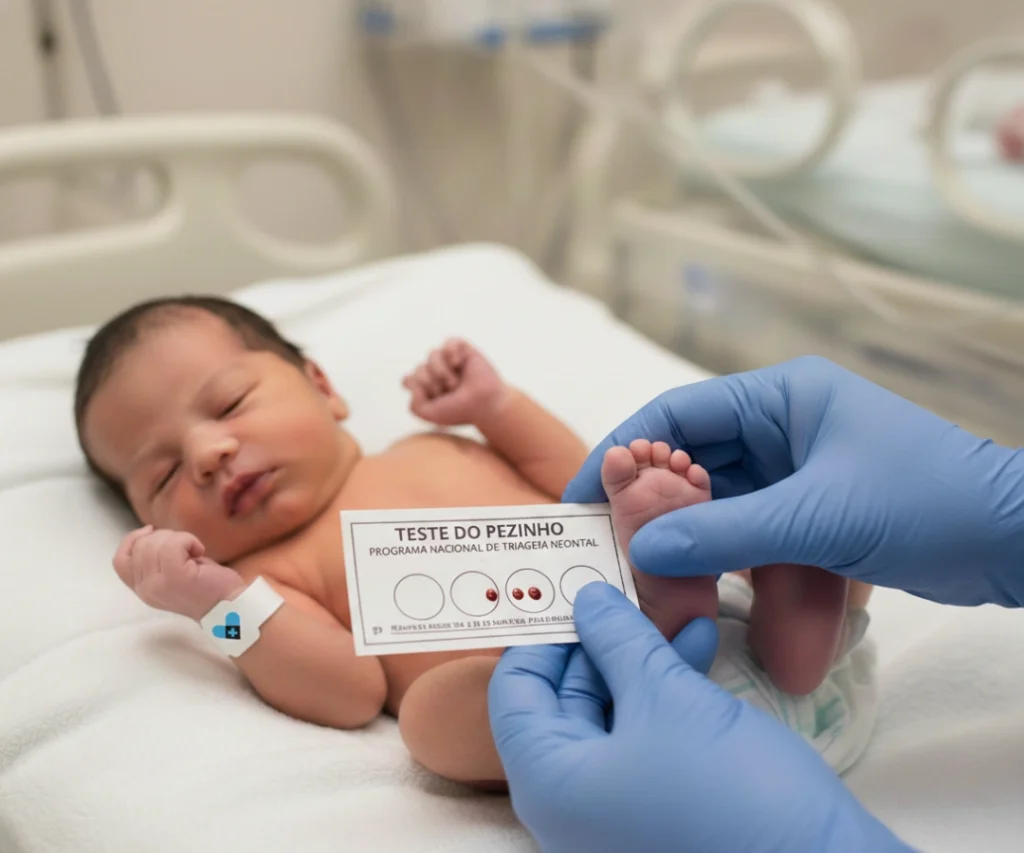
Teste do Pezinho (Triagem Neonatal)
O Teste do Pezinho ampliado é o método mais eficaz para detectar a leucinose precocemente. Ele deve ser realizado entre o 3º e o 5º dia de vida.
Importante: O teste básico do SUS nem sempre inclui a Doença do Xarope de Bordo. A versão ampliada é a mais indicada para identificar doenças metabólicas raras.
Exames confirmatórios
Para confirmar o diagnóstico, eu recomendo exames como:
Dosagem plasmática de aminoácidos
Detecção de níveis elevados de leucina
Análise de cetoácidos (corpos cetônicos) na urina
Teste genético molecular
Avaliação metabólica especializada
Tratamento da Doença do Xarope de Bordo
A leucinose não tem cura, mas pode ser controlada com tratamento rigoroso e acompanhamento contínuo.
1. Dieta restritiva de proteínas
Limitação rigorosa de alimentos ricos em proteínas naturais:
Carnes
Ovos
Leite
Leguminosas
Grãos integrais
2. Fórmulas metabólicas especiais
Leites e suplementos sem leucina, isoleucina e valina
Garantem nutrição adequada sem intoxicação metabólica
3. Monitoramento laboratorial frequente
Dosagem regular de aminoácidos no sangue
Ajustes constantes na dieta
4. Manejo de crises metabólicas
Em situações de infecção ou estresse físico, eu destaco a importância de internação hospitalar e suporte metabólico imediato.
Hidratação intravenosa
Controle rigoroso de aminoácidos
Nutrição emergencial
5. Transplante hepático (em casos selecionados)
Pode melhorar significativamente o controle metabólico
Reduz o risco de crises graves
Indicado apenas em situações específicas
Prognóstico e Qualidade de Vida
Com diagnóstico precoce e adesão ao tratamento:
Crianças podem ter crescimento normal
Desenvolvimento intelectual pode ser preservado
Redução significativa de complicações neurológicas
Vida escolar e social ativa
Sem tratamento adequado:
Alto risco de sequelas neurológicas graves
Comprometimento motor e cognitivo
Risco de mortalidade precoce
Cuidados Essenciais para Famílias e Pacientes
Acompanhamento com médico geneticista
Suporte de nutricionista metabólico
Monitoramento laboratorial regular
Educação alimentar rigorosa
Planejamento genético familiar
Rede de apoio psicológico
Conclusão
A Doença do Xarope de Bordo (Leucinose) é uma condição genética rara, mas potencialmente grave. Com base na experiência clínica e nas evidências científicas, reforço que o diagnóstico precoce é o fator mais determinante para preservar a função neurológica e a qualidade de vida.
Investir em triagem neonatal ampliada, educação familiar e acompanhamento contínuo pode prevenir sequelas neurológicas e salvar vidas.
Perguntas Frequentes
Sim. Embora seja mais comum em recém-nascidos, formas leves ou intermitentes da leucinose podem ser diagnosticadas apenas na adolescência ou na vida adulta, especialmente após crises metabólicas.
O diagnóstico definitivo é feito por análise dos aminoácidos plasmáticos associada ao teste genético molecular para identificação de mutações no complexo BCKD.
Sim. Se não tratada precocemente, o acúmulo de leucina pode causar danos neurológicos permanentes e prejuízo cognitivo.
Sim, desde que haja acompanhamento médico e nutricional, pois exercícios intensos podem aumentar o risco de descompensação metabólica.
Carnes, ovos, leite integral, leguminosas e alimentos ricos em proteínas devem ser rigorosamente controlados para evitar intoxicação metabólica.
Sim. Casais portadores podem realizar aconselhamento genético e testes pré-natais para avaliar o risco em futuras gestações.
O transplante hepático não é considerado cura genética, mas pode normalizar o metabolismo dos aminoácidos e reduzir drasticamente o risco de crises metabólicas.
Sonolência intensa, vômitos, irritabilidade, perda de apetite, odor adocicado acentuado na urina e piora neurológica são sinais precoces de alerta.
Sim. Ela faz parte do grupo dos erros inatos do metabolismo, que inclui outras doenças hereditárias que afetam enzimas metabólicas.
Sim. A doença não impede a fertilidade, mas recomenda-se aconselhamento genético para avaliar o risco de transmissão.
- STRAUSS, K. A.; MORTON, D. H. Maple Syrup Urine Disease. GeneReviews. University of Washington, Seattle, 2021. Disponível em: https://www.ncbi.nlm.nih.gov/books/NBK1319/ . Acesso em: 15 jan. 2026.
- BLACKBURN, P. R. et al. Maple Syrup Urine Disease: Mechanisms and Management. Journal of Inherited Metabolic Disease, v. 40, n. 4, p. 505–516, 2017. Disponível em: https://doi.org/10.1007/s10545-017-0042-9 . Acesso em: 25 jan. 2026.
- ZSCHOCKE, J.; HOFFMANN, G. F. Vademecum Metabolicum: Diagnosis and Treatment of Inborn Errors of Metabolism. 4. ed. Stuttgart: Thieme, 2011.
- BURLINA, A. et al. Inborn Errors of Metabolism in the Neonate: Diagnosis and Treatment. Journal of Pediatrics, v. 161, n. 2, p. 283–289, 2012. Disponível em: https://doi.org/10.1016/j.jpeds.2012.01.038 . Acesso em: 22 jan. 2026.
- WALTER, J. H.; LEE, P. J.; BURTON, B. K. Disorders of Branched Chain Amino Acid Metabolism. Orphanet Journal of Rare Diseases, v. 9, n. 33, 2014. Disponível em: https://doi.org/10.1186/1750-1172-9-33 . Acesso em: 29 jan. 2026.
- VAN CALCAR, S. C. et al. Nutrition Management of Maple Syrup Urine Disease. Molecular Genetics and Metabolism, v. 117, n. 2, p. 102–111, 2016. Disponível em: https://doi.org/10.1016/j.ymgme.2015.11.012 . Acesso em: 29 jan. 2026.
- SUMMONS, R. E.; SASS, J. O.; RINALDO, P. Expanded Newborn Screening for MSUD. Clinical Chemistry, v. 56, n. 7, p. 1239–1246, 2010. Disponível em: https://doi.org/10.1373/clinchem.2010.144154 . Acesso em: 23 jan. 2026.
- NELSON, D. L.; COX, M. M. Lehninger Principles of Biochemistry. 7. ed. New York: W. H. Freeman, 2017.

Lincoln Soledade, – Biomédico CRBM 41556/ES. Mestrando em Gestão em Saúde Pública. Especialista em Patologia Clínica e pós-graduado em Estética Avançada, atua desde 2018 na área de diagnóstico clínico hospitalar. Ao longo de sua trajetória, tem se dedicado à aplicação rigorosa do conhecimento científico para promover diagnósticos precisos, contribuindo significativamente para a qualidade do cuidado em saúde.
